Report of GSEA results for NSG-CDX-BR16, NSG-LM2 models and patient data
Francesc Castro-Giner
2022-02-23
Last updated: 2022-05-12
Checks: 7 0
Knit directory:
diamantopoulou-ctc-dynamics/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220425) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version a747daf. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: analysis/0_differential_expression_gsea_gsva.md
Untracked: analysis/about.md
Untracked: analysis/br16_dge.md
Untracked: analysis/br16_pca.md
Untracked: analysis/core_gene_sets.md
Untracked: analysis/gsea_across_models.md
Untracked: analysis/index.md
Untracked: analysis/license.md
Untracked: analysis/patients_ctc_counts_distribution.md
Untracked: data/differential_expression/
Untracked: data/patients/
Untracked: data/resources/
Untracked: data/sce/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/gsea_across_models.Rmd)
and HTML (docs/gsea_across_models.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | a747daf | fcg-bio | 2022-05-12 | Updated legenf title |
| html | 1fc87b5 | fcg-bio | 2022-05-10 | Build site. |
| html | 8fb5513 | fcg-bio | 2022-05-10 | Build site. |
| html | 8acaa64 | fcg-bio | 2022-04-26 | Build site. |
| Rmd | 545ee28 | fcg-bio | 2022-04-26 | release v1.0 |
| html | 545ee28 | fcg-bio | 2022-04-26 | release v1.0 |
| html | 74b1891 | fcg-bio | 2022-04-26 | Build site. |
| html | c0865c6 | fcg-bio | 2022-04-26 | Build site. |
| html | a136590 | fcg-bio | 2022-04-26 | Build site. |
| html | bfb622b | fcg-bio | 2022-04-26 | Build site. |
| Rmd | 2ef6f7f | fcg-bio | 2022-04-26 | added gsea and gsva tables |
| html | 1006c84 | fcg-bio | 2022-04-26 | Build site. |
| Rmd | 0ded9f5 | fcg-bio | 2022-04-26 | added final code |
Load libraries, additional functions and data
Setup environment
knitr::opts_chunk$set(results='asis', echo=TRUE, message=FALSE, warning=FALSE, error=FALSE, fig.align = 'center', fig.width = 3.5, fig.asp = 0.618, dpi = 600, dev = c("png", "pdf"), fig.showtext = TRUE)
options(stringsAsFactors = FALSE)Load packages
library(tidyverse)
library(showtext)
library(scater)
library(clusterProfiler)
library(enrichplot)
library(ComplexHeatmap)
library(circlize)
library(RColorBrewer)
library(cowplot)
library(DT)
library(GSVA)
library(limma)
library(colorblindr)
library(ggbeeswarm)Set font family for figures
font_add("Helvetica", "./configuration/fonts/Helvetica.ttc")
showtext_auto()Load ggplot theme
source("./configuration/rmarkdown/ggplot_theme.R")Load color palettes
source("./configuration/rmarkdown/color_palettes.R")Load functions
source('./code/R-functions/gse_report.r')
clean_msigdb_names <- function(x) x %>% gsub('REACTOME_', '', .) %>% gsub('WP_', '', .) %>% gsub('BIOCARTA_', '', .) %>% gsub('KEGG_', '', .) %>% gsub('PID_', '', .) %>% gsub('GOBP_', '', .) %>% gsub('_', ' ', .)Load MSigDB gene sets
gmt_files_symbols <- list(
msigdb.c2.cp = './data/resources/MSigDB/v7.4/c2.cp.v7.4.symbols.gmt',
msigdb.c5.bp = './data/resources/MSigDB/v7.4/c5.go.bp.v7.4.symbols.gmt'
)
gmt_files_entrez <- list(
msigdb.c2.cp = './data/resources/MSigDB/v7.4/c2.cp.v7.4.entrez.gmt',
msigdb.c5.bp = './data/resources/MSigDB/v7.4/c5.go.bp.v7.4.entrez.gmt'
)
# combine MSigDB.C2.CP and GO:BP
msigdb.c2.cp_file <- gsub('c2.cp', 'c2.cp.c5.bp', gmt_files_symbols$msigdb.c2.cp)
if(!file.exists(msigdb.c2.cp_file)) {
cat_cmd <- paste('cat', gmt_files_symbols$msigdb.c5.bp, gmt_files_symbols$msigdb.c2.cp, '>',msigdb.c2.cp_file)
system(cat_cmd)
}
gmt_files_symbols$msigdb.c2.cp.c5.bp <- msigdb.c2.cp_file
gmt_sets <- lapply(gmt_files_symbols, function(x) read.gmt(x) %>% collect %>% .[['term']] %>% levels)Load results from differential gene expression analyses
dge_lm2 <- readRDS(file.path(params$dge_dir, 'lm2', 'dge_edgeR_QLF_robust.rds'))
dge_patient <- readRDS(file.path(params$dge_dir, 'patient', 'dge_edgeR_QLF_robust.rds'))Load GSEA results
gse_gsea_br16 <- readRDS(file.path(params$dge_dir, 'br16', 'gse_gsea.rds'))Load LM2 timekinetics data
sce_lm2tk <- readRDS(file.path(params$sce_dir, 'sce_lm2_tk.rds'))
gsva_lm2tk <- readRDS(file.path(params$dge_dir, 'lm2_tk', 'gsva_c2.cp.c5.bp.rds'))NSG-CDX-BR16 GSEA results
Gene set enrichment analysis from differentially expressed genes in CTCs of NSG-CDX-BR16 mice during the rest phase versus active phase.Table listing the enriched gene sets (n = 138, adjusted P value < 0.05) in CTCs obtained in rest versus active phase from NSG-CDX-BR16 mice. The gene set enrichment analysis (GSEA) was performed using ranking genes as input, according to fold-change as shown in Supplementary table 2. P values were obtained using FGSEA method with an eps parameter of 1e-10.
gse_gsea_br16$GSEA$msigdb.c2.cp.c5.bp@result %>%
filter(p.adjust < 0.05) %>%
dplyr::select(ID, setSize, enrichmentScore, NES, pvalue, p.adjust, leading_edge, core_enrichment) %>%
mutate(
NES = round(NES, 2),
pvalue = format.pval(pvalue, digits = 2),
p.adjust = format.pval(p.adjust, digits = 2)
) %>%
rename(
`Term ID` = ID,
`Set size` = setSize,
`Enrichment score` = enrichmentScore,
`P value` = pvalue,
`Adjusted P value` = p.adjust,
`Leading edge` = leading_edge,
`Core enrichment` = core_enrichment
) %>%
datatable(.,
rownames = FALSE,
filter = 'top',
caption = 'Gene set enrichment analysis from differentially expressed genes in CTCs of NSG-CDX-BR16 mice during the rest phase versus active phase.',
extensions = 'Buttons',
options = list(
dom = 'Blfrtip',
buttons = c('csv', 'excel')
))NSG-CDX-BR16 : similarity matrix for enriched gene sets
Generate the data for the similarity heatmap
use_gse_res <- gse_gsea_br16$GSEA$msigdb.c2.cp.c5.bp
# Number of terms to show
showCategoryN <- 30
# Calculate jaccard simialrity index
use_gse_res <- pairwise_termsim(use_gse_res, method = 'JC')
# Collect sim matrix for top N terms
use_terms <- use_gse_res@result %>%
filter(p.adjust < 0.001) %>% head(showCategoryN) %>% collect %>% .[['ID']]
use_mat <- use_gse_res@termsim[use_terms,use_terms]
# Collect results for selected terms
use_res <- use_gse_res@result[use_terms,]
# Transform matrix to symmetric
for(x in rownames(use_mat)){
for(y in colnames(use_mat)) {
if(x == y) {
use_mat[x,y] <- 1
} else {
max_sim <- max(c(use_mat[x,y], use_mat[y,x]), na.rm = TRUE)
use_mat[x,y] <- max_sim
use_mat[y,x] <- max_sim
}
}
}
# Collect FC values for ridge plot. Values are capped at -2 and 2
gs2id <- geneInCategory(use_gse_res)[seq_len(showCategoryN)]
gs2val <- lapply(gs2id, function(id) {
res <- use_gse_res@geneList[id]
res <- res[!is.na(res)]
})
gs2val_capped <- lapply(gs2val, function(x) {x[x > 2] <- 2; x[x < -2] <- -2; x} )
lt = lapply(gs2val_capped, function(x) data.frame(density(x)[c("x", "y")]))
# Save matrix for future use
br16_gsea_sim_mat <- use_matGenerate row annotation
nes_colors <- c(
brewer.pal(n = 7, name ="BrBG")[6],
brewer.pal(n = 7, name ="BrBG")[2]
)
ha_row_nes = rowAnnotation(
NES = anno_barplot(
use_res$NES,
baseline = 0,
width = unit(1, "cm"),
bar_width = 0.7,
gp = gpar(
fill = ifelse(use_res$NES < 0 , nes_colors[1], nes_colors[2]),
col = ifelse(use_res$NES < 0 , nes_colors[1], nes_colors[2])
)
),
annotation_name_gp = gpar(fontsize = 8)
)
col_fun_nes = colorRamp2(
seq(max(use_res$NES), min(use_res$NES), length.out = 8),
brewer.pal(n = 8, name ="BrBG") %>% rev)
ha_row_nes_ht = rowAnnotation(
NES = use_res$NES,
border = c( NES = TRUE),
col = list( NES = col_fun_nes),
simple_anno_size = unit(0.8, "cm"),
annotation_name_rot = 0,
annotation_name_gp = gpar(fontsize = 8)
)
col_fun_pval = colorRamp2(
seq(max(-log10(use_res$p.adjust)), -log10(0.05), length.out = 8),
brewer.pal(n = 8, name ="Reds") %>% rev)
ha_row_pval = rowAnnotation(
`-log10\n(adjusted\np value)` = -log10(use_res$p.adjust),
border = c( `-log10\n(adjusted\np value)` = TRUE),
col = list( `-log10\n(adjusted\np value)` = col_fun_pval),
simple_anno_size = unit(0.8, "cm"),
annotation_name_rot = 0,
annotation_name_gp = gpar(fontsize = 8),
annotation_legend_param = list(title_gp = gpar(fontsize = 8),labels_gp = gpar(fontsize = 8))
)Similarity matrix without row names
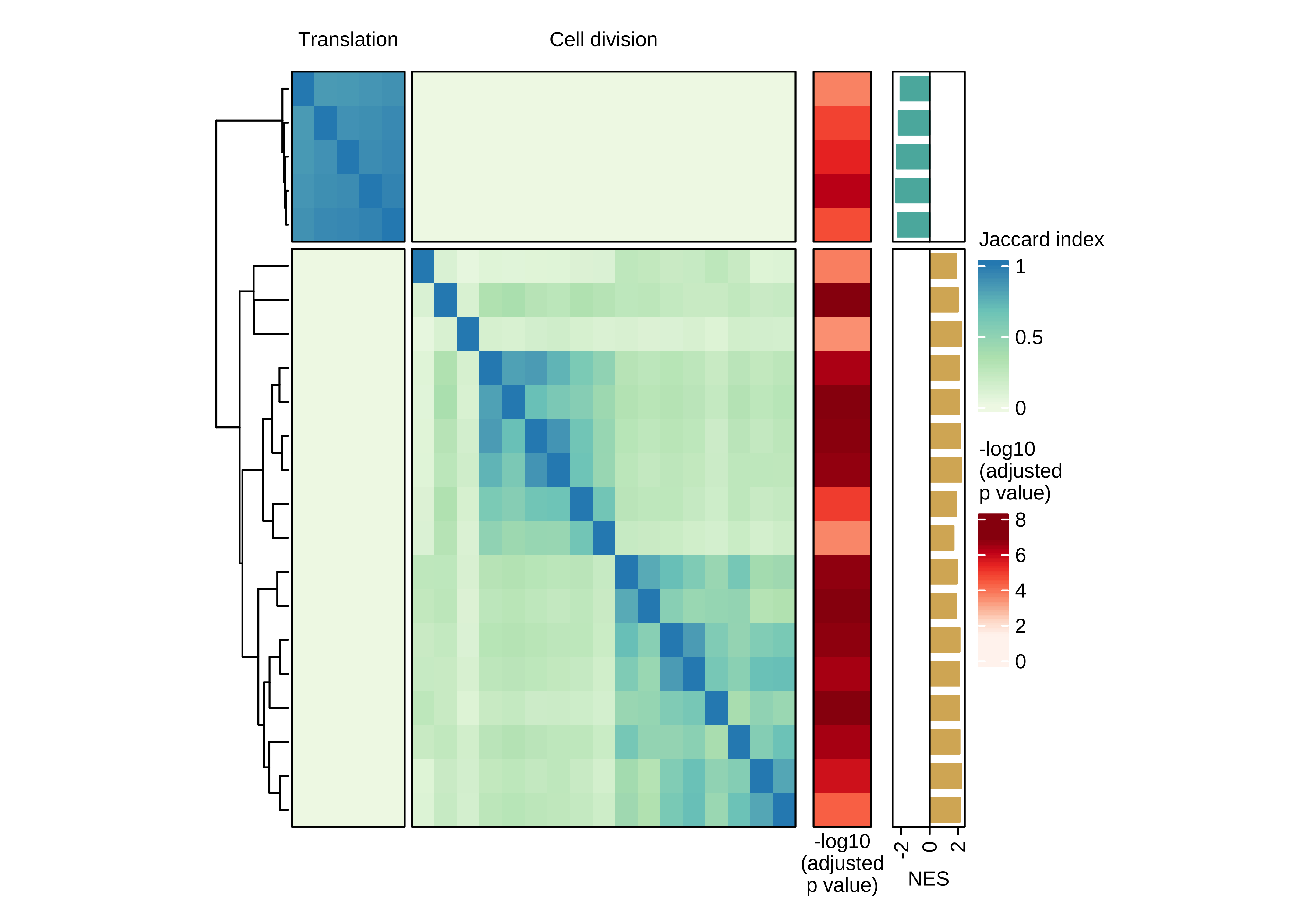
Heatmap showing the pair-wise similarity matrix of enriched gene sets (gene set enrichment analysis (GSEA), adjusted P value ≤ 0.0001) using differential expression between CTCs of rest and active phase from NSG-CDX-BR16 mice. Heatmap colors represent the Jaccard similarity coefficient. The heatmap on the right represents the adjusted P value as obtained in GSEA.
col_fun <- colorRamp2(seq(0, 1, length.out = 4), brewer.pal(4, "GnBu"))
n_split <- 2
ha_top <- HeatmapAnnotation(
foo = anno_block(
labels = c("Translation", "Cell division"),
labels_gp = gpar(col = "black", fontsize = 8),
gp = gpar(lwd = 0, lty = 0))
)
ht <- Heatmap(
use_mat,
name = 'Jaccard index',
column_split = n_split,
row_split = n_split,
column_title = NULL,
row_title = NULL,
col = col_fun,
show_column_dend = FALSE,
show_column_names = FALSE,
border = TRUE,
top_annotation = ha_top,
heatmap_legend_param = list(title_gp = gpar(fontsize = 8),labels_gp = gpar(fontsize = 8)),
width = unit(7, "cm"))
ht_br16_c2 <- draw(ht + ha_row_pval + ha_row_nes, ht_gap = unit(c(0.2, 0.3, 0.3), "cm"))
for (slice in 1:n_split) {
decorate_annotation("NES", {
grid.lines(unit(c(0, 0), "native"), unit(c(0, 1), "npc"), gpar(lty = 2))
}, slice = slice)
}
| Version | Author | Date |
|---|---|---|
| 1006c84 | fcg-bio | 2022-04-26 |
cat("\n\n")Similarity matrix with row names
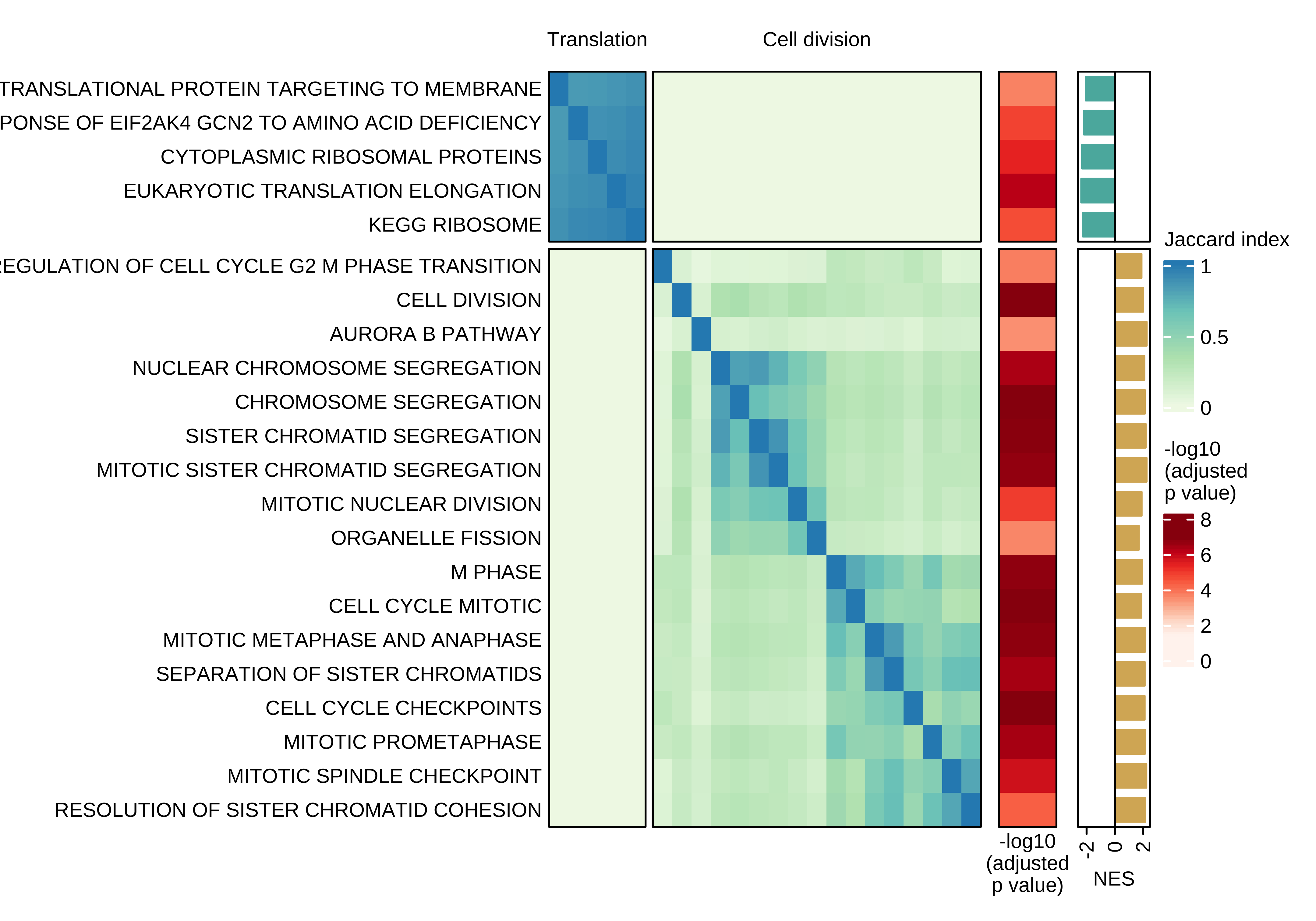
Heatmap showing the pair-wise similarity matrix of enriched gene sets (gene set enrichment analysis (GSEA), adjusted P value ≤ 0.0001) using differential expression between CTCs of rest and active phase from NSG-CDX-BR16 mice. Heatmap colors represent the Jaccard similarity coefficient. The heatmap on the right represents the adjusted P value as obtained in GSEA.
use_mat_rn <- use_mat
rownames(use_mat_rn) <- rownames(use_mat_rn) %>%
gsub("REACTOME_", "", .) %>%
gsub("BIOCARTA_", "", .) %>%
gsub("^PID_", "", .) %>%
gsub("^WP_", "", .) %>%
gsub("^PID_", "", .) %>%
gsub("^GOBP_", "", .) %>%
gsub("_", " ", .)
ht <- Heatmap(
use_mat_rn,
name = 'Jaccard index',
column_split = n_split,
row_split = n_split,
column_title = NULL,
row_title = NULL,
col = col_fun,
show_column_dend = FALSE,
show_column_names = FALSE,
show_row_dend = FALSE,
row_names_side = "left",
row_names_gp = gpar(fontsize = 8),
row_names_max_width = unit(7, "cm"),
border = TRUE,
top_annotation = ha_top,
heatmap_legend_param = list(title_gp = gpar(fontsize = 8),
labels_gp = gpar(fontsize = 8)
),
width = unit(6, "cm"))
draw(ht + ha_row_pval + ha_row_nes, ht_gap = unit(c(0.2, 0.3, 0.3), "cm"))
for (slice in 1:n_split) {
decorate_annotation("NES", {
grid.lines(unit(c(0, 0), "native"), unit(c(0, 1), "npc"), gpar(lty = 2))
}, slice = slice)
}
| Version | Author | Date |
|---|---|---|
| 1006c84 | fcg-bio | 2022-04-26 |
cat("\n\n")Save selected gsets for future use
gse_gsea_br16_f <- gse_gsea_br16$GSEA$msigdb.c2.cp.c5.bp@result %>% filter(p.adjust < 0.001)
row_order <- row_order(ht_br16_c2) %>% unlist
use_gsets <- rownames(br16_gsea_sim_mat)[row_order]
use_gmt_gsets <- read.gmt(gmt_files_symbols$msigdb.c2.cp.c5.bp)
use_gmt_gsets <- use_gmt_gsets %>% filter(term %in% use_gsets)
saveRDS(use_gsets, file = file.path(params$dge_dir, 'br16', 'ht_br16_c2_gene_sets.Rmd'))GSEA for NSG-LM2 and Patient
Run GSEA using candidate pathways from NSG-CDX-BR16.
NSG-LM2
output_dir <- file.path(params$dge_dir, 'lm2')
dge <- dge_lm2
fc_list <- dge$results$logFC %>% set_names(dge$results$gene_name) %>% sort(decreasing = TRUE)
gsea_lm2 <- GSEA(fc_list, TERM2GENE=use_gmt_gsets, pvalueCutoff = 1)
gsea_lm2 <- pairwise_termsim(gsea_lm2)Patient
output_dir <- file.path(params$dge_dir, 'patient')
dge <- dge_patient
fc_list <- dge$results$logFC %>% set_names(dge$results$gene_name) %>% sort(decreasing = TRUE)
gsea_patient <- GSEA(fc_list, TERM2GENE=use_gmt_gsets, pvalueCutoff = 1)
gsea_patient <- pairwise_termsim(gsea_patient)Combine NSG-CDX-BR16, NSG-LM2 and patient GSEA data
gsea_lm2@result$donor <- 'LM2'
gsea_patient@result$donor <- 'Patient'
gsea_br16 <- gse_gsea_br16_f %>%
mutate(donor = 'Br16') %>%
dplyr::select(one_of(colnames(gsea_lm2@result)))
gsea_comb <- rbind(gsea_br16, gsea_lm2@result, gsea_patient@result)
gsea_comb <- gsea_comb %>%
left_join(gse_gsea_br16_f %>% dplyr::select(ID, NES, p.adjust),
by = 'ID',
suffix = c("", ".br16")) %>%
mutate(ID = factor(ID, levels = rev(use_gsets)))Table differential expression in NSG-LM2
Genes differentially expressed in CTCs of NSG-LM26 mice during the rest phase versus active phase. Table listing the differentially expressed genes comparing CTCs obtained in the rest phase (n = 65) versus the active phase (n = 73) of NSG-CDX-BR16 mice. All genes evaluated are included in the table (n = 12,261). Fold-change and P values were computed with the quasi-likelihood (QL) approach from edgeR using robust dispersion estimates. For fold-change calculation, active phase samples were used in the denominator.
dge_lm2$results %>%
dplyr::select(gene_name, gene_type, logFC, logCPM, PValue, FDR, description) %>%
rownames_to_column('Ensemble ID') %>%
mutate(
logFC = round(logFC, 2),
logCPM = round(logCPM, 2),
PValue = format.pval(PValue, digits = 2),
FDR = format.pval(FDR, digits = 2),
description = gsub(" \\[.*\\]", "", description)
) %>%
dplyr::rename(
`Gene name` = gene_name,
`Gene type` = gene_type
) %>%
datatable(.,
rownames = FALSE,
filter = 'top',
caption = 'Differential expression analysis in CTCs of NSG-LM2 mice during the rest phase versus active phase.',
extensions = 'Buttons',
options = list(
dom = 'Blfrtip',
buttons = c('csv', 'excel')
))Table GSEA in NSG-LM2
Gene set enrichment analysis from gene expression in CTCs of NSG-LM2 mice during the rest phase versus active phase. Table listing the gene set enrichment results in CTCs obtained in rest versus active phase from NSG-LM2 mice. Only gene sets enriched in NSG-CDX-BR16 were analysed (n = 22, adjusted P value ≤ 0.0001). The gene set enrichment analysis (GSEA) was performed using ranked genes by fold-change as input.
gsea_lm2@result %>%
dplyr::select(ID, setSize, enrichmentScore, NES, pvalue, p.adjust, leading_edge, core_enrichment) %>%
mutate(
NES = round(NES, 2),
pvalue = format.pval(pvalue, digits = 2),
p.adjust = format.pval(p.adjust, digits = 2)
) %>%
rename(
`Term ID` = ID,
`Set size` = setSize,
`Enrichment score` = enrichmentScore,
`P value` = pvalue,
`Adjusted P value` = p.adjust,
`Leading edge` = leading_edge,
`Core enrichment` = core_enrichment
) %>%
datatable(.,
rownames = FALSE,
filter = 'top',
caption = 'Gene set enrichment analysis in CTCs of NSG-LM2 mice during the rest phase versus active phase. Only enriched gene sets from NSG-CDX-BR16 were analysed.',
extensions = 'Buttons',
options = list(
dom = 'Blfrtip',
buttons = c('csv', 'excel')
))Table differential expression in Patients
Genes differentially expressed in CTCs of breast cancer patients during the rest phase versus active phase. Table listing the differentially expressed genes comparing CTCs obtained in the rest phase (n = 65) versus the active phase (n = 73) of NSG-CDX-BR16 mice. All genes evaluated are included in the table (n = 12,261). Fold-change and P values were computed with the quasi-likelihood (QL) approach from edgeR using robust dispersion estimates. For fold-change calculation, active phase samples were used in the denominator.
dge_patient$results %>%
dplyr::select(gene_name, gene_type, logFC, logCPM, PValue, FDR, description) %>%
rownames_to_column('Ensemble ID') %>%
mutate(
logFC = round(logFC, 2),
logCPM = round(logCPM, 2),
PValue = format.pval(PValue, digits = 2),
FDR = format.pval(FDR, digits = 2),
description = gsub(" \\[.*\\]", "", description)
) %>%
dplyr::rename(
`Gene name` = gene_name,
`Gene type` = gene_type
) %>%
datatable(.,
rownames = FALSE,
filter = 'top',
caption = 'Differential expression analysis in CTCs of breast cancer patient during the rest phase versus active phase.',
extensions = 'Buttons',
options = list(
dom = 'Blfrtip',
buttons = c('csv', 'excel')
))Table GSEA in Patients
Gene set enrichment analysis from gene expression in CTCs of breast cancer patients during the rest phase versus active phase. Table listing the gene set enrichment results in CTCs obtained in rest versus active phase from breast cancer patients. Only gene sets enriched in NSG-CDX-BR16 were analysed (n = 22, adjusted P value ≤ 0.0001). The gene set enrichment analysis (GSEA) was performed using ranked genes by fold-change as input.
gsea_patient@result %>%
dplyr::select(ID, setSize, enrichmentScore, NES, pvalue, p.adjust, leading_edge, core_enrichment) %>%
mutate(
NES = round(NES, 2),
pvalue = format.pval(pvalue, digits = 2),
p.adjust = format.pval(p.adjust, digits = 2)
) %>%
rename(
`Term ID` = ID,
`Set size` = setSize,
`Enrichment score` = enrichmentScore,
`P value` = pvalue,
`Adjusted P value` = p.adjust,
`Leading edge` = leading_edge,
`Core enrichment` = core_enrichment
) %>%
datatable(.,
rownames = FALSE,
filter = 'top',
caption = 'Gene set enrichment analysis in CTCs of breast cancer patient during the rest phase versus active phase. Only enriched gene sets from NSG-CDX-BR16 were analysed.',
extensions = 'Buttons',
options = list(
dom = 'Blfrtip',
buttons = c('csv', 'excel')
))GSEA for NSG-CDX-BR16 and NSG-LM2
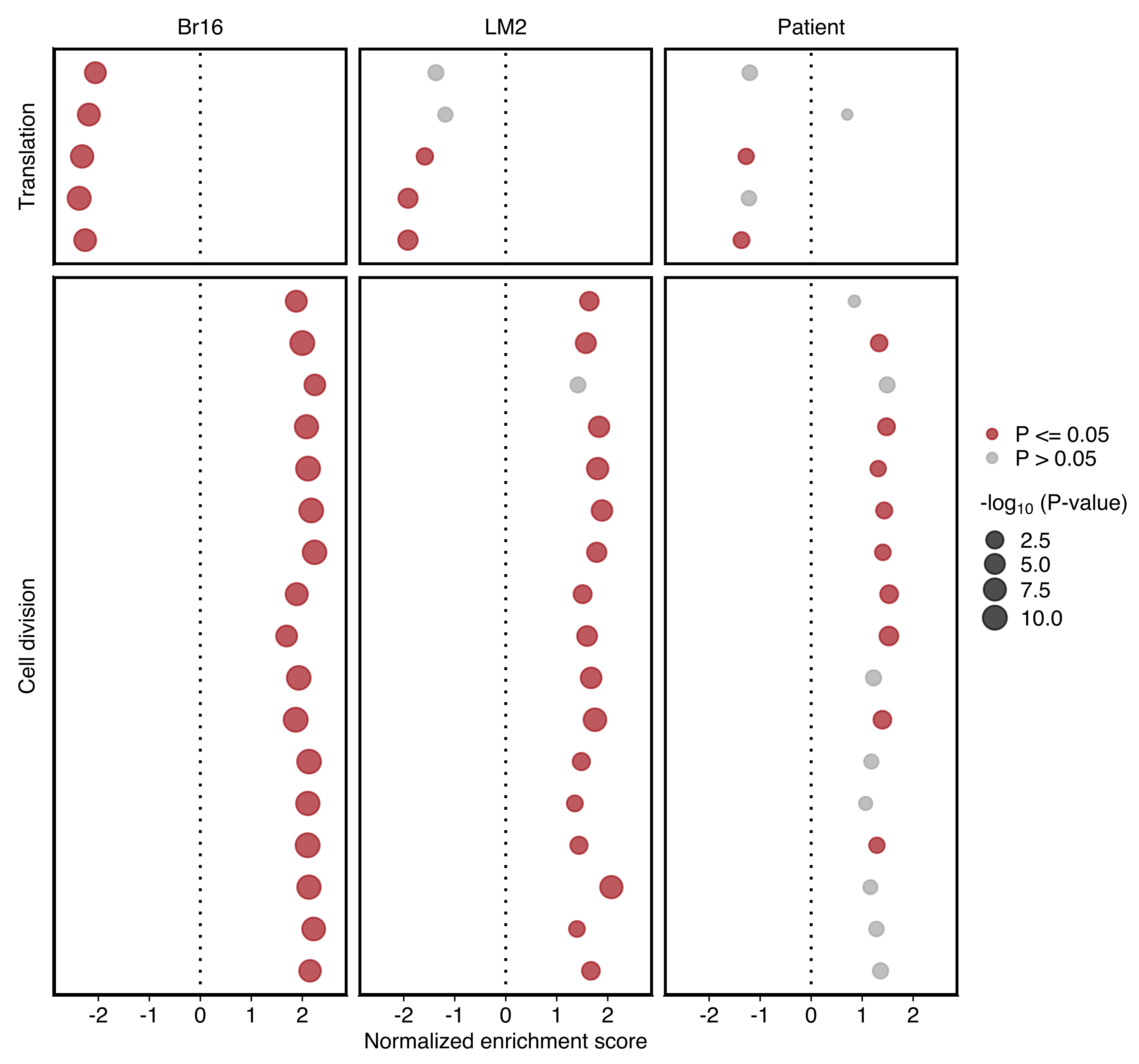
Plot comparing the normalized enrichment score (NES) and adjusted P value (dot size) obtained using GSEA for gene sets shown in “d”. Left and right panels show the results for NSG-CDX-BR16 and NSG-LM2 models, respectively. Gene sets with an adjusted P value ≤ 0.05 in each sample set are highlighted in red.
label_func <- default_labeller(18)
xlim <- (gsea_comb$NES %>% abs %>% max) + 0.25
dotplot_br16 <- gsea_comb %>%
filter(donor == 'Br16') %>%
mutate(
color = ifelse(pvalue <= 0.05, 'P <= 0.05', 'P > 0.05'),
row_split = ifelse(NES.br16 < 0, 'Translation', 'Cell division') %>% factor(levels=c('Translation', 'Cell division'))
) %>%
ggplot(aes(x = NES, y = ID, size = -log10(pvalue), color = color)) +
geom_point(alpha = 0.7) +
scale_color_manual(values = c(`P <= 0.05` = 'firebrick', `P > 0.05` = 'grey70')) +
scale_y_discrete(labels = label_func) +
scale_size(range = c(1.5, 3.8)) +
labs(
x = 'Normalized enrichment score',
y = NULL,
color = NULL,
size = bquote("-log"[10] ~ .(paste0("(P-value)")))
) +
facet_grid(cols = vars(donor), row = vars(row_split), scales = 'free_y', space = 'free', switch = "y") +
xlim(c(-xlim, xlim)) +
geom_vline(xintercept = 0, lty = 3) +
panel_border(color = "black") +
theme(
axis.title.y=element_blank(),
axis.ticks.y=element_blank(),
axis.text.y=element_blank(),
strip.background = element_rect(fill = 'white'),
strip.placement = "outside"
)
dotplot_lm2<- gsea_comb %>%
filter(donor == 'LM2') %>%
mutate(
color = ifelse(pvalue <= 0.05, 'P <= 0.05', 'P > 0.05'),
row_split = ifelse(NES.br16 < 0, 'Translation', 'Cell division') %>% factor(levels=c('Translation', 'Cell division'))
) %>%
ggplot(aes(x = NES, y = ID, size = -log10(pvalue), color = color)) +
geom_point(alpha = 0.7) +
scale_color_manual(values = c(`P <= 0.05` = 'firebrick', `P > 0.05` = 'grey70')) +
scale_y_discrete(labels = label_func) +
scale_size(range = c(1.5, 3.8)) +
labs(
x = 'Normalized enrichment score',
y = NULL,
color = NULL,
size = bquote("-log"[10] ~ .(paste0("(P-value)")))
) +
facet_grid(cols = vars(donor), row = vars(row_split), scales = 'free_y', space = 'free', switch = "y") +
xlim(c(-xlim, xlim)) +
geom_vline(xintercept = 0, lty = 3) +
panel_border(color = "black") +
theme(
axis.title.y=element_blank(),
axis.ticks.y=element_blank(),
axis.text.y=element_blank(),
strip.background = element_rect(fill = 'white'),
strip.placement = "outside"
)
plot_grid(dotplot_br16, dotplot_lm2)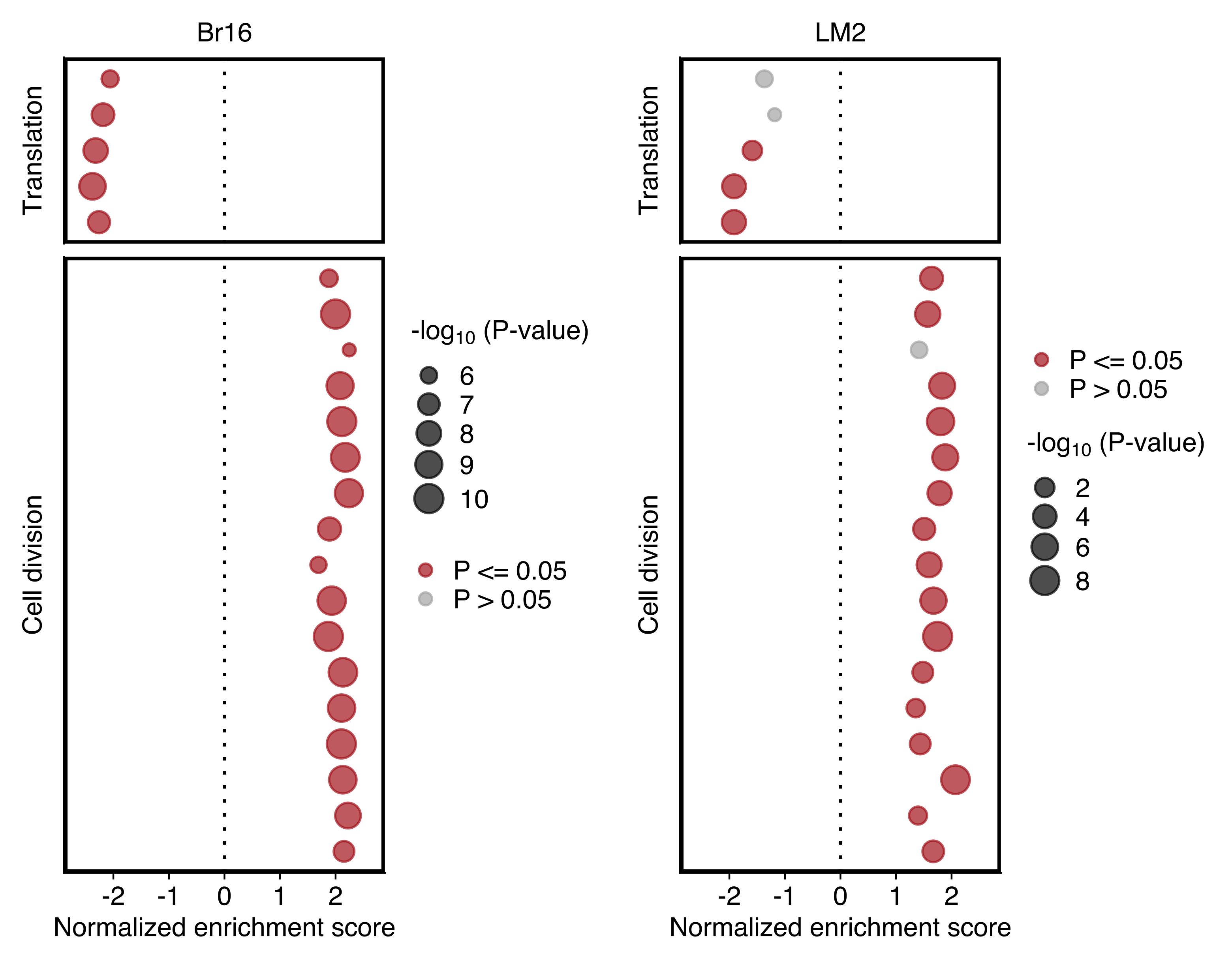
| Version | Author | Date |
|---|---|---|
| 1006c84 | fcg-bio | 2022-04-26 |
GSEA for Patient
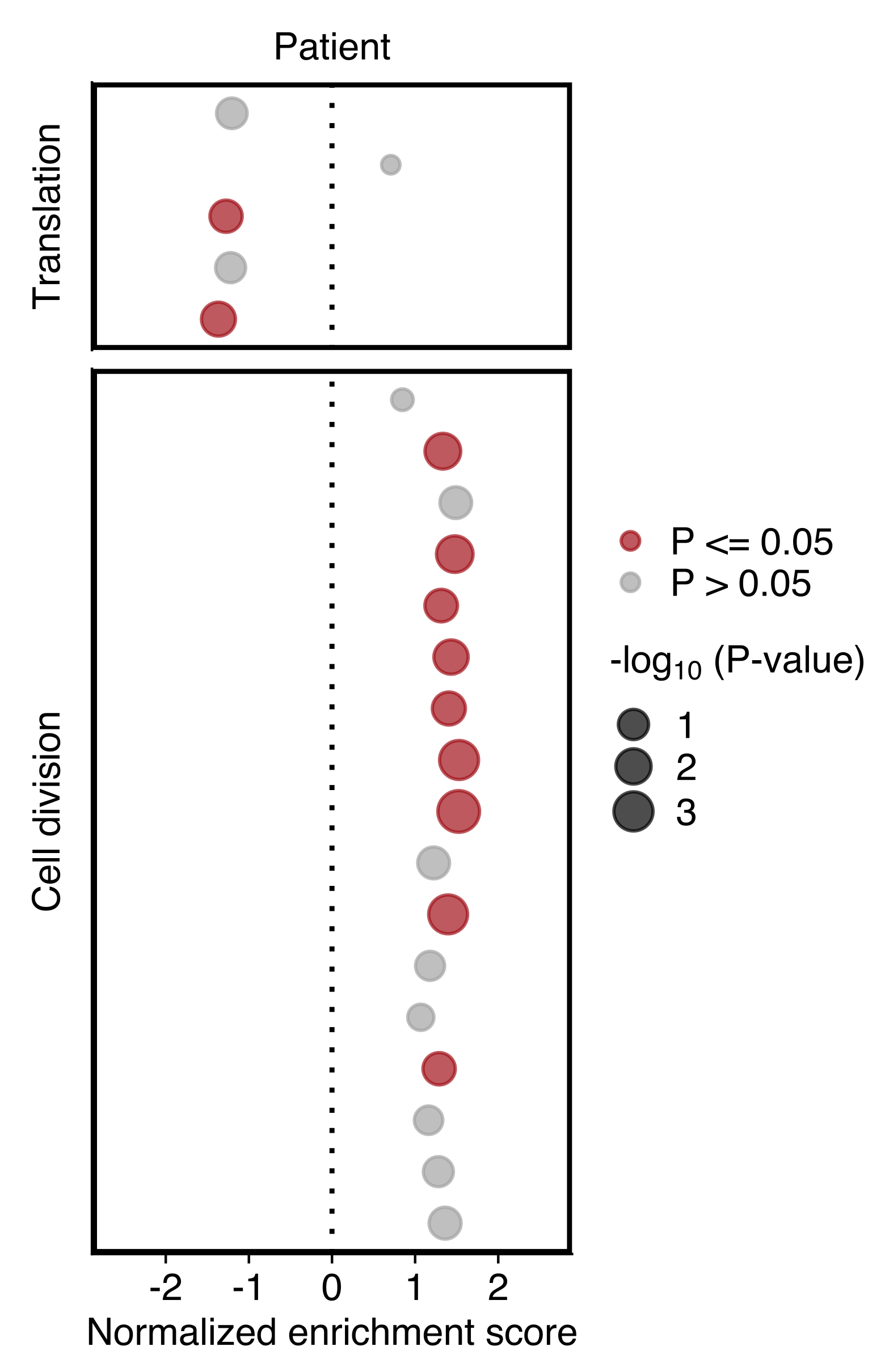
Plot showing the NES and P value (dot size) in patient CTCs obtained using GSEA for gene sets shown in “d”. Gene sets with an P value ≤ 0.05 are highlighted in red (bottom).
label_func <- default_labeller(18)
xlim <- (gsea_comb$NES %>% abs %>% max) + 0.25
gsea_comb %>%
filter(donor == 'Patient') %>%
mutate(
color = ifelse(pvalue <= 0.05, 'P <= 0.05', 'P > 0.05'),
row_split = ifelse(NES.br16 < 0, 'Translation', 'Cell division') %>% factor(levels=c('Translation', 'Cell division'))
) %>%
ggplot(aes(x = NES, y = ID, size = -log10(pvalue), color = color)) +
geom_point(alpha = 0.7) +
scale_color_manual(values = c(`P <= 0.05` = 'firebrick', `P > 0.05` = 'grey70')) +
scale_y_discrete(labels = label_func) +
scale_size(range = c(1.5, 3.8)) +
labs(
x = 'Normalized enrichment score',
y = NULL,
color = NULL,
size = bquote("-log"[10] ~ .(paste0("(P-value)")))
) +
facet_grid(cols = vars(donor), row = vars(row_split), scales = 'free_y', space = 'free', switch = "y") +
xlim(c(-xlim, xlim)) +
geom_vline(xintercept = 0, lty = 3) +
panel_border(color = "black") +
theme(
axis.title.y=element_blank(),
axis.ticks.y=element_blank(),
axis.text.y=element_blank(),
strip.background = element_rect(fill = 'white'),
strip.placement = "outside"
)
| Version | Author | Date |
|---|---|---|
| 1006c84 | fcg-bio | 2022-04-26 |
GSEA for NSG-CDX-BR16, NSG-LM2 and patient
Plots comparing the normalized enrichment score (NES) and adjusted P value (dot size) obtained using GSEA for gene sets shown in “d”. Gene sets with an adjusted P value ≤ 0.05 in each sample set are highlighted in red
label_func <- default_labeller(18)
xlim <- (gsea_comb$NES %>% abs %>% max) + 0.25
gsea_comb %>%
mutate(
color = ifelse(pvalue <= 0.05, 'P <= 0.05', 'P > 0.05'),
row_split = ifelse(NES.br16 < 0, 'Translation', 'Cell division') %>% factor(levels=c('Translation', 'Cell division'))
) %>%
ggplot(aes(x = NES, y = ID, size = -log10(pvalue), color = color)) +
geom_point(alpha = 0.7) +
scale_color_manual(values = c(`P <= 0.05` = 'firebrick', `P > 0.05` = 'grey70')) +
scale_y_discrete(labels = label_func) +
scale_size(range = c(1.5, 3.8)) +
labs(
x = 'Normalized enrichment score',
y = NULL,
color = NULL,
size = bquote("-log"[10] ~ .(paste0("(P-value)")))
) +
facet_grid(cols = vars(donor), row = vars(row_split), scales = 'free_y', space = 'free', switch = "y") +
xlim(c(-xlim, xlim)) +
geom_vline(xintercept = 0, lty = 3) +
panel_border(color = "black") +
theme(
axis.title.y=element_blank(),
axis.ticks.y=element_blank(),
axis.text.y=element_blank(),
strip.background = element_rect(fill = 'white'),
strip.placement = "outside"
)
| Version | Author | Date |
|---|---|---|
| 1006c84 | fcg-bio | 2022-04-26 |
GSVA for NSG-LM2
Run differential expression
Run differential expression at pathway level removing timepoint 0600 (ZT = 0, only 1 biological replicate) and using only candidate pathways from BR16 analysis. We use limma (Smyth 2004) as suggested in GSVA vignette. For several groups (timepoints) we are using the strategy defined at limma vignette Section 9.3
use_sce <- sce_lm2tk
gsva_res <- gsva_lm2tk
use_gsets_nes <- gse_gsea_br16$GSEA$msigdb.c2.cp.c5.bp@result[use_gsets, 'NES'] %>% set_names(use_gsets)
use_gsets_cat <- ifelse(use_gsets_nes < 0, 'Translation', 'Cell division') %>% factor(levels=c('Translation', 'Cell division'))
use_gmt_gsets <- read.gmt(gmt_files_symbols$msigdb.c2.cp.c5.bp)
use_gmt_gsets <- use_gmt_gsets %>% filter(term %in% use_gsets)
# Remove 06000 samples, only one replicate
use_samples <- intersect(colnames(gsva_res), use_sce[,use_sce$timepoint!='0600']$sample_alias)
# limma
f <- use_sce[,use_samples]$timepoint %>% factor
design <- model.matrix(~ 0 + f)
gsva_res_sel <- gsva_res[use_gsets,use_samples]
fit <- lmFit(gsva_res_sel, design)
contrast_to_eval <- combn(colnames(design), 2, simplify = TRUE) %>% apply(., 2, function(x) paste(x, collapse = '-'))
contrast.matrix <- makeContrasts(contrasts = contrast_to_eval, levels = colnames(design))
fit2 <- contrasts.fit(fit, contrast.matrix)
fit2 <- eBayes(fit2)
limma_res <- topTable(fit2, number=100000) %>%
rownames_to_column('Term') %>%
mutate(to_rownames = Term) %>%
column_to_rownames('to_rownames')
limma_res$Term_cat <- use_gsets_cat[limma_res$Term]Additional objects for plotting
coldata_ord <- colData(use_sce) %>% data.frame %>% arrange(zt, sample_type)
gsva_mat <- gsva_res[use_gsets, coldata_ord$sample_alias]
gsva_df <- gsva_mat %>% data.frame %>%
rownames_to_column('term') %>%
pivot_longer(-term, names_to = 'sample_alias') %>%
left_join(coldata_ord) %>%
mutate(term = factor(term, levels = use_gsets))
gsva_df$term_cat <- use_gsets_cat[gsva_df$term]
gsva_avg_df <- gsva_df %>%
group_by(zt, timepoint, term) %>%
summarise(mean_gsva = mean(value)) %>%
mutate(term = factor(term, levels = use_gsets))
gsva_avg_df$term_cat <- use_gsets_cat[gsva_avg_df$term]
gsva_df$term <- clean_msigdb_names(gsva_df$term) %>% factor(., clean_msigdb_names(use_gsets))
gsva_avg_df$term <- clean_msigdb_names(gsva_avg_df$term) %>% factor(., clean_msigdb_names(use_gsets))
gsva_avg_mat <- gsva_avg_df %>%
ungroup() %>%
dplyr::select(-term_cat, -timepoint) %>%
pivot_wider(names_from = zt, values_from = mean_gsva) %>%
column_to_rownames('term') %>%
as.matrix
gsva_avg_mat <- gsva_avg_mat[clean_msigdb_names(use_gsets),]GSVA table of results
Gene set variation analysis in CTCs of theNSG-LM2 time-kinetics experiment. Table listing the results from the differential analysis of gene set variation analysis (GSVA) scores in CTC of the NSG-LM2 time-kinetics experiment. Only gene sets enriched in NSG-CDX-BR16 were analysed (n = 22, adjusted P value ≤ 0.0001). The F statistic and the corresponding P value generated by limma, combine the pair-wise comparisons between all the time points in the experiment with more than three replicates (n = 6 comparisons). GSVA scores for each individual sample are listed at the end of the table.
gsva_res_sel %>%
as.data.frame %>%
rownames_to_column('Term') %>%
left_join(limma_res) %>%
mutate(
P.Value = format.pval(P.Value, digits = 2),
adj.P.Val = format.pval(adj.P.Val, digits = 2)
) %>%
dplyr::select(Term, Term_cat,AveExpr:adj.P.Val, LM2_Clusters_0200_1:LM2_WBC_2200_1) %>%
rename(
`Term ID` = Term,
`Category` = Term_cat,
`Average GSVA score` = AveExpr,
`P value` = P.Value,
`Adjusted P value` = adj.P.Val,
) %>%
datatable(.,
rownames = FALSE,
filter = 'top',
caption = 'Results from differential enrichment across timepoints of NSG-LM2 time kinetics experiment. Only enriched gene sets from NSG-CDX-BR16 were reported. The F statistic and the corresponding P value combine the pair-wise comparisons between all the time points in the experiment with more than three replicates. GSVA scores for each individual sample are listed at the end of the table.',
extensions = 'Buttons',
options = list(
dom = 'Blfrtip',
buttons = c('csv', 'excel')
))GSVA across time series
GSVA score for translation (yellow, n= 5) and cell division (blue, n= 17) gene sets in CTCs obtained from the NSG-LM2 time-kinetics experiment. Yellow and blue lines represent the average at each time point. Individual points represent the enrichment score for each CTC sample. The white and grey backgrounds represent environmental light (rest period) and dark conditions (active period), respectively. Differential expression adjusted P values as obtained from limma are shown for each individual gene set.
bg_color <- data.frame(
xmin = c(-2, 0, 12),
xmax = c(0, 12, 22),
fill_bg = c('night', 'day', 'night')
)
adj_p_df <- limma_res %>%
mutate(
term = clean_msigdb_names(limma_res$Term) %>% factor(., clean_msigdb_names(use_gsets)),
label = format.pval(adj.P.Val, digits = 1),
label = paste("italic('P=')~", label),
ypos = ifelse(Term_cat == 'Translation', -0.45, 0.45)
)
ggplot() +
geom_rect(data = bg_color, aes(xmin = xmin, xmax = xmax, ymin = -Inf, ymax = Inf, fill = fill_bg), alpha = 0.5) +
geom_hline(yintercept = 0, lty = 2, size = 0.2) +
geom_quasirandom(data = gsva_df, aes(zt, value, group = term, color = term_cat), size = 1, pch = 16, alpha = 0.4, width = 0.3) +
geom_line(data = gsva_avg_df, aes(zt, mean_gsva, group = term, color = term_cat), size = 0.6, alpha = 1) +
facet_wrap(~term, labeller = label_wrap_gen(width = 25), ncol = 5, scales = 'free_x') +
scale_fill_manual(values = c('night' = "grey80", 'day' = "white")) +
scale_color_OkabeIto() +
labs(
x = 'Time (ZT)',
y = 'GSVA enrichment score',
color = NULL,
fill = NULL
) +
guides(fill = FALSE) +
scale_x_continuous(
expand = c(0,0),
breaks=c(0, 4, 12, 16, 20)
) +
scale_y_continuous(
expand = c(0,0),
limits = c(-0.55, 0.55)
) +
theme(
legend.position="top",
plot.margin = margin(14, 7, 3, 1.5),
strip.background = element_rect(fill = 'white'),
strip.text = element_text(size = 6)
) +
geom_text(x = 16, aes(label = label, y = ypos), data = adj_p_df, size = 1.8, hjust = 0, parse = TRUE)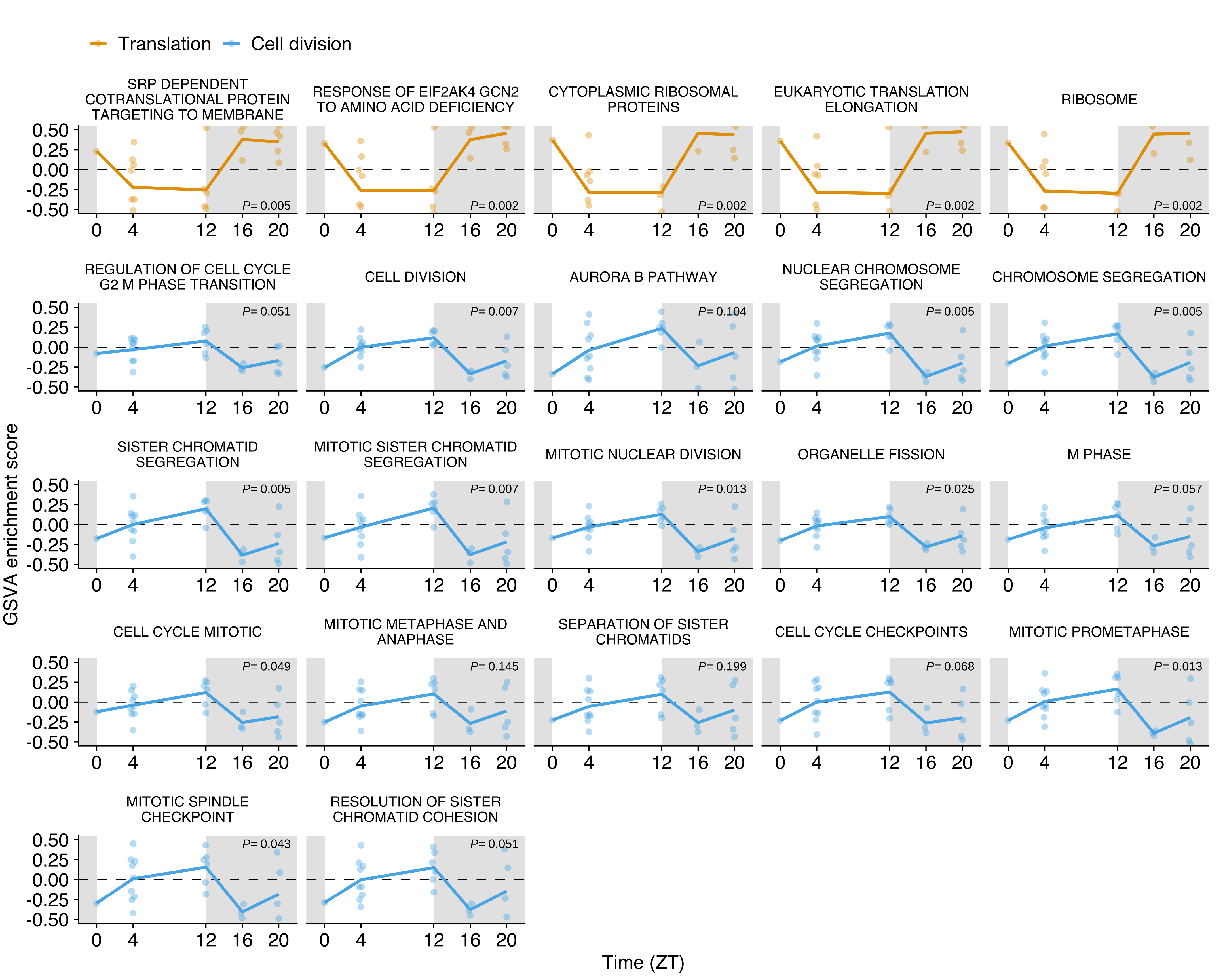
| Version | Author | Date |
|---|---|---|
| 1006c84 | fcg-bio | 2022-04-26 |
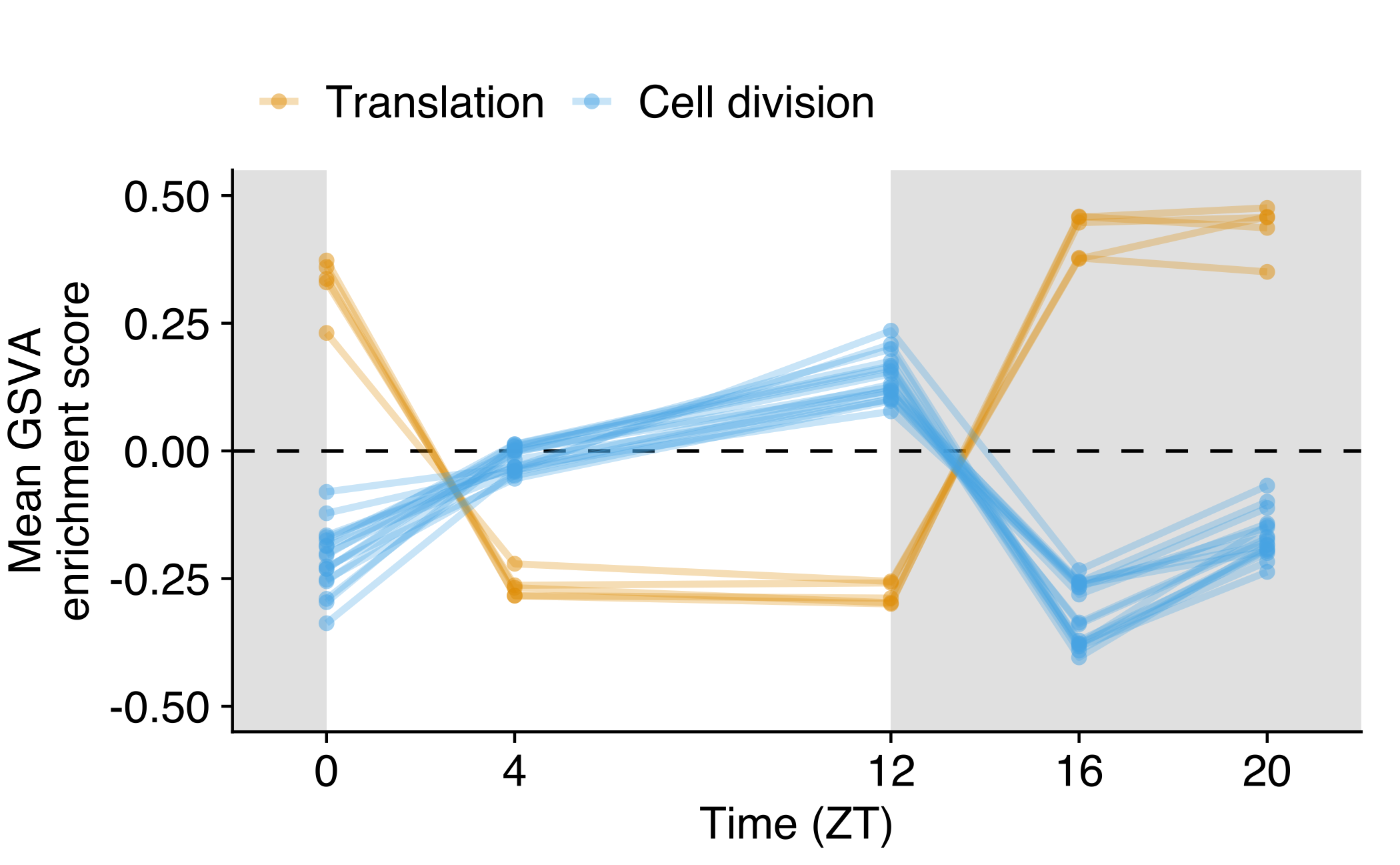
Average GSVA score across time series
Average GSVA score for translation (yellow, n=5) and cell division (blue, n=17) gene sets in CTCs obtained in the NSG-LM2 time-kinetics experiment. The average was calculated for each gene set and time point across all CTC samples (ZT0 n=1, ZT4 n=9, ZT12 n=6, ZT16 n=3, ZT20 n=5). The white and grey backgrounds represent environmental light (rest period) and dark conditions (active period), respectively.
bg_color <- data.frame(
xmin = c(-2, 0, 12),
xmax = c(0, 12, 22),
fill_bg = c('night', 'day', 'night')
)
ggplot() +
geom_rect(data = bg_color, aes(xmin = xmin, xmax = xmax, ymin = -Inf, ymax = Inf, fill = fill_bg), alpha = 0.5) +
geom_hline(yintercept = 0, lty = 2, size = 0.3) +
geom_line(data = gsva_avg_df, aes(zt, mean_gsva, group = term, color = term_cat), size = 0.6, alpha = 0.3) +
geom_point(data = gsva_avg_df, aes(zt, mean_gsva, group = term, color = term_cat), size = 1, pch = 16, alpha = 0.5) +
scale_fill_manual(values = c('night' = "grey80", 'day' = "white")) +
scale_color_OkabeIto() +
labs(
x = 'Time (ZT)',
y = 'Mean GSVA\nenrichment score',
color = NULL,
fill = NULL
) +
guides(fill = "none") +
scale_x_continuous(
expand = c(0,0),
breaks=c(0, 4, 12, 16, 20)
) +
scale_y_continuous(
expand = c(0,0),
limits = c(-0.55, 0.55)
) +
theme(
legend.position="top",
plot.margin = margin(14, 7, 3, 1.5)
)
| Version | Author | Date |
|---|---|---|
| 1006c84 | fcg-bio | 2022-04-26 |
sessionInfo()R version 4.1.0 (2021-05-18) Platform: x86_64-apple-darwin17.0 (64-bit) Running under: macOS Big Sur 10.16
Matrix products: default BLAS: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRblas.dylib LAPACK: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRlapack.dylib
locale: [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages: [1] grid parallel stats4 stats graphics
grDevices utils
[8] datasets methods base
other attached packages: [1] ggbeeswarm_0.6.0 colorblindr_0.1.0
[3] colorspace_2.0-2 limma_3.48.3
[5] GSVA_1.40.1 DT_0.19
[7] cowplot_1.1.1 RColorBrewer_1.1-2
[9] circlize_0.4.13 ComplexHeatmap_2.8.0
[11] enrichplot_1.12.3 clusterProfiler_4.0.5
[13] scater_1.20.1 scuttle_1.2.1
[15] SingleCellExperiment_1.14.1 SummarizedExperiment_1.22.0 [17]
Biobase_2.52.0 GenomicRanges_1.44.0
[19] GenomeInfoDb_1.28.4 IRanges_2.26.0
[21] S4Vectors_0.30.2 BiocGenerics_0.38.0
[23] MatrixGenerics_1.4.3 matrixStats_0.61.0
[25] showtext_0.9-4 showtextdb_3.0
[27] sysfonts_0.8.5 forcats_0.5.1
[29] stringr_1.4.0 dplyr_1.0.7
[31] purrr_0.3.4 readr_2.0.2
[33] tidyr_1.1.4 tibble_3.1.5
[35] ggplot2_3.3.5 tidyverse_1.3.1
[37] workflowr_1.6.2
loaded via a namespace (and not attached): [1] utf8_1.2.2
tidyselect_1.1.1
[3] htmlwidgets_1.5.4 RSQLite_2.2.8
[5] AnnotationDbi_1.54.1 BiocParallel_1.26.2
[7] scatterpie_0.1.7 munsell_0.5.0
[9] ScaledMatrix_1.0.0 codetools_0.2-18
[11] withr_2.4.2 GOSemSim_2.18.1
[13] highr_0.9 knitr_1.36
[15] rstudioapi_0.13 DOSE_3.18.3
[17] labeling_0.4.2 git2r_0.28.0
[19] GenomeInfoDbData_1.2.6 polyclip_1.10-0
[21] bit64_4.0.5 farver_2.1.0
[23] rhdf5_2.36.0 rprojroot_2.0.2
[25] downloader_0.4 vctrs_0.3.8
[27] treeio_1.16.2 generics_0.1.1
[29] xfun_0.27 R6_2.5.1
[31] doParallel_1.0.16 clue_0.3-60
[33] graphlayouts_0.7.1 rsvd_1.0.5
[35] rhdf5filters_1.4.0 bitops_1.0-7
[37] cachem_1.0.6 fgsea_1.18.0
[39] gridGraphics_0.5-1 DelayedArray_0.18.0
[41] assertthat_0.2.1 promises_1.2.0.1
[43] scales_1.1.1 ggraph_2.0.5
[45] beeswarm_0.4.0 gtable_0.3.0
[47] beachmat_2.8.1 Cairo_1.5-12.2
[49] tidygraph_1.2.0 rlang_0.4.12
[51] GlobalOptions_0.1.2 splines_4.1.0
[53] lazyeval_0.2.2 broom_0.7.10
[55] yaml_2.2.1 reshape2_1.4.4
[57] modelr_0.1.8 crosstalk_1.1.1
[59] backports_1.3.0 httpuv_1.6.3
[61] qvalue_2.24.0 tools_4.1.0
[63] ggplotify_0.1.0 ellipsis_0.3.2
[65] jquerylib_0.1.4 Rcpp_1.0.7
[67] plyr_1.8.6 sparseMatrixStats_1.4.2
[69] zlibbioc_1.38.0 RCurl_1.98-1.5
[71] GetoptLong_1.0.5 viridis_0.6.2
[73] cluster_2.1.2 haven_2.4.3
[75] ggrepel_0.9.1 fs_1.5.0
[77] magrittr_2.0.1 data.table_1.14.2
[79] DO.db_2.9 reprex_2.0.1
[81] whisker_0.4 xtable_1.8-4
[83] hms_1.1.1 patchwork_1.1.1
[85] evaluate_0.14 XML_3.99-0.8
[87] readxl_1.3.1 shape_1.4.6
[89] gridExtra_2.3 compiler_4.1.0
[91] crayon_1.4.2 shadowtext_0.0.9
[93] htmltools_0.5.2 ggfun_0.0.4
[95] later_1.3.0 tzdb_0.2.0
[97] aplot_0.1.1 lubridate_1.8.0
[99] DBI_1.1.1 tweenr_1.0.2
[101] dbplyr_2.1.1 MASS_7.3-54
[103] Matrix_1.3-4 cli_3.1.0
[105] igraph_1.2.7 pkgconfig_2.0.3
[107] xml2_1.3.2 foreach_1.5.1
[109] annotate_1.70.0 ggtree_3.0.4
[111] vipor_0.4.5 bslib_0.3.1
[113] XVector_0.32.0 rvest_1.0.2
[115] yulab.utils_0.0.4 digest_0.6.28
[117] graph_1.70.0 Biostrings_2.60.2
[119] rmarkdown_2.11 cellranger_1.1.0
[121] fastmatch_1.1-3 tidytree_0.3.5
[123] GSEABase_1.54.0 DelayedMatrixStats_1.14.3 [125] rjson_0.2.20
lifecycle_1.0.1
[127] nlme_3.1-153 jsonlite_1.7.2
[129] Rhdf5lib_1.14.2 BiocNeighbors_1.10.0
[131] viridisLite_0.4.0 fansi_0.5.0
[133] pillar_1.6.4 lattice_0.20-45
[135] KEGGREST_1.32.0 fastmap_1.1.0
[137] httr_1.4.2 GO.db_3.13.0
[139] glue_1.4.2 png_0.1-7
[141] iterators_1.0.13 bit_4.0.4
[143] HDF5Array_1.20.0 ggforce_0.3.3
[145] stringi_1.7.5 sass_0.4.0
[147] blob_1.2.2 BiocSingular_1.8.1
[149] memoise_2.0.0 irlba_2.3.3
[151] ape_5.5